Prothèses mammaires : la réglementation sur la sellette
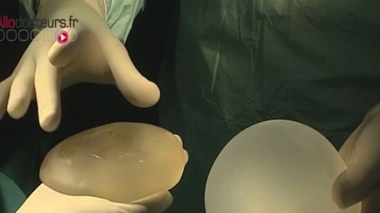
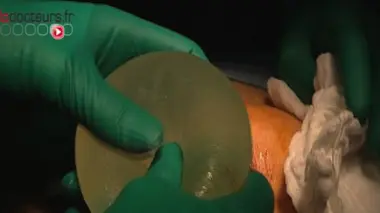
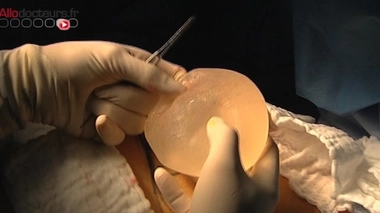
Le scandale des implants mammaires PIP a mis en lumière une faille dans la réglementation des dispositifs médicaux. Contrairement aux médicaments, ces matériels et équipements destinés au diagnostic, à la prévention, au contrôle, au traitement ou encore à l’atténuation d’une maladie ou d’une blessure, ne sont pas soumis à une procédure d’autorisation de mise sur le marché.
Par La rédaction d'Allo Docteurs
Mis à jour le
Seringues, défibrillateurs cardiaques, fauteuils roulants, pansements ou implants mammaires… Contrairement aux médicaments, les dispositifs médicaux ne sont pas soumis à une procédure d’autorisation. Leur mise sur le marché est régie par la directive européenne 93/42/CE d’application obligatoire depuis juin 1998, qui impose aux fabricants de dispositifs médicaux l’apposition d’un marquage CE sur leur produit, avant leur commercialisation. Ce marquage matérialise la conformité du dispositif aux exigences essentielles de santé et de sécurité du produit.
Pour pouvoir apposer le marquage CE, le fabricant doit réaliser des contrôles et des essais qui assurent sa conformité aux exigences européennes de santé et de sécurité. Une fois qu’il est apposé, le pays a le droit de circuler dans tous les pays de l’Union européenne.
Les dispositifs médicaux sont classés en quatre classes en fonction de leur dangerosité. Hormis les dispositifs médicaux les moins à risques (classe 1), la démarche suivie par un fabricant pour démontrer la conformité de ses produits avant la mise sur marché est évaluée par un organisme notifié, choisi par le fabricant dans la liste des organismes désignés par les autorités compétentes de l’Union européenne.
Ce dernier évalue la documentation technique et le système complet d’assurance qualité du fabricant. L’évaluation du système qualité comprend un audit dans les locaux du fabricant. A l’issue de cette évaluation, l’organisme notifié délivre un certificat de conformité qui permet au fabricant de marquer CE ses prothèses et de les mettre sur le marché européen. Par ailleurs, après délivrance du certificat CE, les audits sont répétés périodiquement, généralement annuellement, tout au long de la mise sur le marché des produits.
L’Afssaps - comme ses homologues européens - vérifie a posteriori les conditions de mise sur le marché des dispositifs médicaux et notamment leur conformité à la réglementation. Elle peut notamment inspecter les sites de fabrication.
Mais le scandale des prothèses PIP révèle les limites du système actuel de mise sur le marché pour les dispositifs médicaux. Jeudi 5 janvier 2012, le ministre de la Santé a déclaré qu’il fallait "revoir en profondeur la mise sur le marché, mais aussi les formes de contrôle". Il a encore ajouté "sur les dispositifs médicaux, il nous faut d’autres règles. (…) Le simple label ne me suffit pas".
Indépendamment du scandale PIP, l’Europe est en train de revoir sa directive sur les dispositifs médicaux. Des changements pourraient donc intervenir prochainement.
En savoir plus :