Thalassémie : en manque d'hémoglobine
Chaque année, près de 300.000 enfants dans le monde naissent avec une maladie du sang. Parmi eux, plus d'un tiers souffre de thalassémie, une pathologie due à un défaut de synthèse de l'hémoglobine. Comment se transmet cette maladie ? Y a-t-il plusieurs formes de thalassémie ? Peut-on en guérir ?
Par La rédaction d'Allo Docteurs
Mis à jour le
Thalassémie : définition
Pour comprendre ce qu'est la thalassémie : les explications de Marina et Benoît
En France, on estime qu'entre 400 et 500 personnes sont atteintes de thalassémie. Une maladie du sang génétique qui, comme son nom l'indique, touche essentiellement les personnes issues du pourtour méditerranéen. En grec, thalassa signifie la "mer", et émie le "sang".
La thalassémie se traduit par un défaut de production de l'hémoglobine. L'hémoglobine est une grosse protéine contenue dans les globules rouges, son rôle est de transporter l'oxygène. Elle est constituée de quatre chaînes protéiques : deux chaînes alpha et deux chaînes bêta. Chacune de ces chaînes est fabriquée par un gène différent. Quand la chaîne alpha est anormale, on parle d'alpha-thalassémie. Quand la chaîne bêta est anormale, on parle de bêta-thalassémie.
Dans les deux types de thalassémie, ces chaînes défaillantes rendent l'hémoglobine incapable de remplir ses fonctions. Il en résulte une anémie qui se traduit par une pâleur, une grande fatigabilité, parfois des vertiges et des essoufflements. La gravité des signes est variable selon la maladie. La bêta-thalassémie est la forme la plus courante, elle est également la forme la plus grave.
Cette maladie génétique a une transmission héréditaire. Quand les parents sont tous les deux porteurs sains du gène de la thalassémie, si l'enfant reçoit une seule version du gène malade, il est lui aussi porteur mais il ne développera pas la maladie puisqu'il a reçu aussi un gène normal qui fabrique de la bonne hémoglobine. En revanche, s'il reçoit de ses parents deux versions du gène malade, il sera thalassémique.
La thalassémie : quels traitements ?
La thalassémie d'Ali Sattarpour, 39 ans, a été diagnostiquée à l'âge de 5 mois. Ses parents, sans le savoir, étaient tous les deux des porteurs sains.
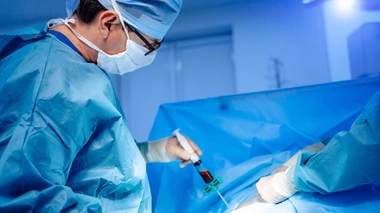
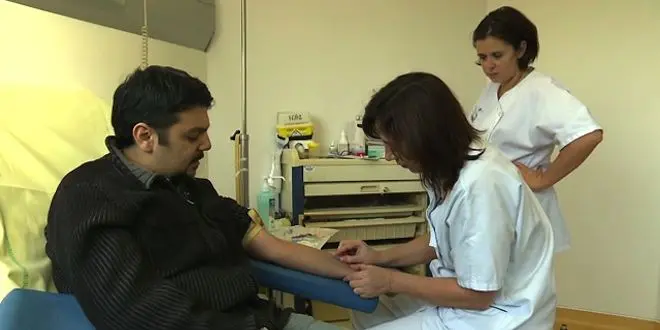
Dans le cas d'une bêta-thalassémie majeure, le patient doit recevoir une transfusion sanguine. Les transfusions sont essentielles pour améliorer le pronostic vital des patients. Une prise de sang est indispensable avant la transfusion. Elle permet de vérifier que le nouveau sang est bien compatible avec celui du patient comme l'explique le Pr France Pirenne, responsable de l'établissement français du sang hôpital Henri-Mondor (AP-HP) : "Pour tous les patients transfusés, on réalise systématiquement une analyse qui s'appelle la RAI. Cet examen a pour but de vérifier si le patient a dans son plasma des anticorps qui pourraient être dirigés contre les globules rouges des poches de sang qu'on va lui transfuser. Et donc si ces anticorps sont présents, on choisira en conséquence les poches de sang pour éviter qu'il y ait des problèmes d'incompatibilité". Et pour plus de sécurité, un second test est effectué.
Mais les transfusions ne sont pas sans conséquences. À chaque transfusion, le corps du patient absorbe une quantité importante de fer qui n'est pas éliminée par les urines ou les selles. À la longue, celui-ci risque de pénétrer dans les cellules des organes et les détruire. C'est pour cela qu'il est vital d'associer à la transfusion un médicament qui élimine la surcharge en fer.
La greffe de moelle osseuse est le seul traitement efficace. Elle permet de relancer la fabrication des globules rouges et donc d'arrêter les transfusions. Malheureusement, elle n'est possible que dans 30% des cas. Autre contrainte, la greffe ne peut réussir que si elle est réalisée à temps, c'est-à-dire avant que la surcharge en fer ne détériore les organes, notamment le foie, la rate et le cœur. Il est donc urgent de trouver d'autres traitements.
Thalassémie et thérapie génique
Thérapie génique : le travail d’un groupe de scientifiques
Un espoir : la thérapie génique. Des équipes scientifiques tentent de trouver une autre solution. Puisque la thalassémie est une maladie génétique due à une mutation (c'est-à-dire qu'une partie du gène a été remplacée par une autre qui est anormale), il suffirait, en théorie, de réparer ce gène malade pour guérir. C'est le principe de la thérapie génique et c'est donc sur cette piste que les chercheurs s'avancent.
Le dépistage. Pour connaître le risque de transmission, il est indispensable de se faire dépister par une simple prise de sang. En cas de grossesse, un diagnostic prénatal est réalisé entre la dixième et la douzième semaine de grossesse.
Thalassémie : l'espoir d'un nouveau traitement
Si la thérapie génique est un traitement intéressant, elle n'est pas dénuée de risques. Depuis deux ans, une équipe de chercheurs de l'Institut Imagine, à Paris, travaille sur une autre piste : celle d'un médicament. Les premiers essais cliniques ont été lancés assez rapidement, il y a un peu plus d'un an. Les travaux continuent mais les premiers résultats sont plutôt spectaculaires.
Grâce à une double injection réalisée une fois par mois, les patients qui ont participé à l'essai clinique ont pu réduire de 30% leurs apports sanguins et donc de fer. Pour les médecins, l'autre bonne nouvelle c'est aussi une amélioration de leur taux d'hémoglobine.
C'est la moelle contenue dans certains de nos os comme le fémur ou les côtes, qui fabrique les plaquettes, les globules blancs, mais aussi les globules rouges. Chez les personnes atteintes de bêta-thalassémie, les globules rouges en devenir sont anormalement nombreux à l'intérieur de la moelle. Faute de chaînes bêta, la plupart meurent avant d'atteindre leur maturité ou présentent un profil anormal et restent incapables de transporter l'oxygène.
Pour résoudre cette énigme, l'équipe du professeur Hermine a tout simplement pris le problème à l'envers : "Dans la bêta-thalassémie, il y a un manque de chaînes bêta. Et on a toujours pensé que si on voulait guérir cette maladie, il fallait ramener la chaîne bêta. Et nous nous sommes aperçus que quand on manque de chaînes bêta, il y a une hyperproduction de chaînes alpha qui est toxique pour la cellule", explique l'hématologue.
En analysant des rates de souris thalassémiques, l'équipe s'est rendue compte que trop de chaînes alpha entraînaient la surproduction d'une protéine. Et cette fameuse protéine toxique que l'équipe a identifié, c'est le GDF11. Le coupable identifié, restait à trouver l'antidote. C'est une autre équipe qui tentait de mettre au point un anti-cancéreux qui leur a apporté sur un plateau. Inefficace contre le cancer, leur molécule augmentait contre toute attente la production de globules rouges. Ce produit est capable d'inhiber la protéine GDF11.
"Nous avons montré qu'en bloquant le GDF11, on était capable de ré-induire une maturation terminale et une fabrication des globules rouges. Cela signifie qu'il y aura plus d'hémoglobine dans le sang des patients, qui seront mieux oxygénés et en meilleure forme. Et tout ce qui faisait pousser les os… va disparaître et les patients se sentiront mieux", raconte le Pr Olivier Hermine. Gagner de l'espérance de vie et moins souffrir pour les 60 patients français qui participent actuellement aux essais, la promesse semble tenue.
L'équipe cherche encore à améliorer son médicament, et elle espère que tous les malades pourront en bénéficier au plus vite. En attendant, pour les 200.000 porteurs sains de bêta-thalassémie en France, il est indispensable en cas de grossesse, de réaliser un diagnostic prénatal entre la dixième et la douzième semaine de grossesse.